


Probes Newsletter 05/2020
Newsletter 05/2020

New Microdeletion Probes from MetaSystems...
Microdeletion syndromes are characterized as heterogenous diseases caused by the loss of small chromosomal regions, typically 1-3 Mb in size - the resulting diseases occur with incidences of up to 1:4.000 in newborns. In general, these genetic aberrations are not visible in classical cytogenetics. Fluorescence in situ hybridization (FISH) is one of the best-established methods to reliably detect microdeletion syndromes and confirm diagnoses of the corresponding diseases.
MetaSystems Probes is expanding its microdeletion portfolio! Two new probes for the detection of Prader-Willi/Angelman (D-5421-050-OG) and Smith-Magenis/Miller-Dieker (D-5422-050-OG) are available now. FISH probes detecting Wolf-Hirschhorn, Cri-du-Chat and Williams-Beuren syndromes have already been available since autumn 2019. For further information, please visit www.metasystems-probes.com.
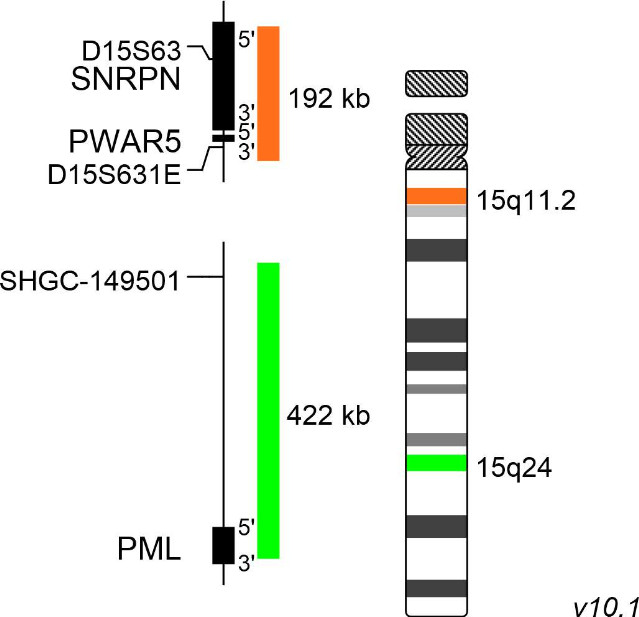
XL Prader-Willi/Angelman
Clinical Applications: Microdeletions
Prader-Willi syndrome (PWS) and Angelman syndrome (AS) are complex neurodevelopmental disorders caused by chromosomal deletion of genes in the 15q11-q13 region. In the most cases, the corresponding genes are silenced on the sister chromosome via genetic imprinting. Heredity of the deleted paternal segment 15q11-q13 leads to development of PWS, while patients carrying the maternal deletion of this segment suffer from AS. The most common genetic cause for PWS is the deletion of the paternal 15q11-q13 copy. Maternal uniparental disomy of 15q11-q13 or translocations with breaks in the 15q11-q13 region are further mechanisms leading to PWS. Genes located in this region, especially SNRPN (small nuclear ribonucleoprotein polypeptide N) and NDN (necdin), are considered to be crucial for disease development. The absence of the maternal UBE3A (ubiquitin-protein ligase E3A) gene copy located in this segment is crucial for the development of AS.
XL Prader-Willi/Angelman detects deletions in the long arm of chromosome 15. The orange labeled probe hybridizes to the SNRPN/PWAR5 locus at 15q11.2. The green labeled probe hybridizes to a specific locus at 15q24 and functions as a reference probe.

XL Smith-Magenis/Miller-Dieker
Clinical Applications: Microdeletions
While interstitial deletions in the segment 17p11.2 are the genetic basis of Smith-Magenis syndrome (SMS), deletions of the more distal part of chromosome 17 (17p13.3) lead to Miller-Dieker syndrome (MDS). The SMS inducing deletions of 17p11.2 have no preferential breakpoints, but most patients analyzed show deletions smaller than 4 Mb. The key gene responsible for the manifestation of the clinical symptoms of SMS is RAI1 (retinoic acid induced 1), coding for a transcription factor. One of the crucial genes involved in the development of Miller-Dieker lissencephaly and craniofacial dysmorphism is the PAFAH1B1 gene encoding the regulatory subunit of the platelet activating factor acetylhydrolase 1b.
XL Smith-Magenis/Miller-Dieker detects deletions in the short arm of chromosome 17. The orange labeled probe hybridizes to the PAFAH1B1 (Miller-Dieker) locus at 17p13. The green labeled probe hybridizes to the RAI1 (Smith-Magenis) gene region at 17p11.2.
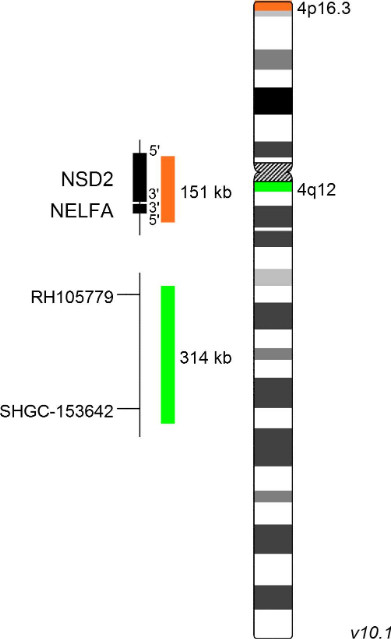
XL Wolf-Hirschhorn
Clinical Applications: Microdeletions
The association of partial 4p16 deletions with Wolf-Hirschhorn syndrome (WHS) was first described by Cooper and Hirschhorn in 1961. Most 4p16 deletions in WHS occur de novo, only in the minority of cases the rearranged chromosome is inherited. WHS is a contiguous gene syndrome associated with haploinsufficiency of several closely linked genes. The severity of clinical manifestation depends on the amount of genetic material affected. Two WHS critical regions (WHSCR) have been identified. The classical WHSCR1, spanning 165 kb on chromosomal location 4p16.3, includes the genes NSD2 (nuclear receptor binding SET domain protein 2) and NELFA (negative elongation factor complex member A). WHSCR2 was defined based on the identification of patients with atypical forms of WHS showing no deletion of WHSCR1. WHSCR2 is located distally to WHSCR1 partially including the gene NSD2, but excluding NELFA.
XL Wolf-Hirschhorn detects deletions in the short arm of chromosome 4. The orange labeled probe hybridizes to the NSD2 (WHSC1) locus at 4p16.3. The green labeled probe hybridizes to a specific locus at 4q12 and functions as a reference probe.
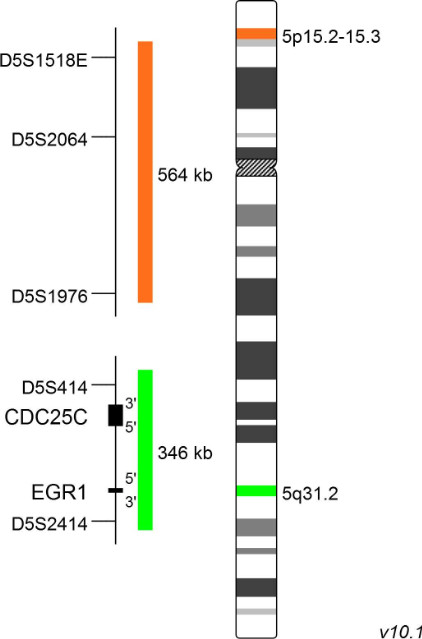
XL Cri-Du-Chat
Clinical Applications: Microdeletions
The Cri-du-Chat syndrome (CdCS), or 5p-minus syndrome, was first described by Lejeune et al in 1963. The name refers to the main clinical feature of the syndrome, a characteristic cat-like cry in early childhood. The majority of patients carry a terminal deletion of the short arm of chromosome 5 with breakpoints ranging from 5p13 to 5p15.2 with a size of up to 40 Mb. Most 5p deletions occur de novo, probably during spermatogenesis. Breakpoints are not well defined and differ between CdCS cases. Only few patients have an interstitial deletion, translocations or other less common aberrations. Patient studies established a link between the size of the deleted region and the CdCS phenotype and identified regions 5p15.2 and 5p15.3 responsible for dysmorphism, mental retardation and the cat-like cry.
XL Cri-Du-Chat detects deletions in the short arm of chromosome 5. The orange labeled probe hybridizes to a chromosomal locus at 5p15.2-15.3. The green labeled probe hybridizes to a specific locus at 5q31.2 and functions as a reference probe.

XL Williams-Beuren
Clinical Applications: Microdeletions
The Williams-Beuren syndrome (WBS) is a rare contiguous gene deletion syndrome. WBS phenotype is complex, age-dependent and varies between individuals. One of the most fatal and characteristic clinical complications is supravalvular aortic stenosis (SVAS). The WBS is associated with a chromosomal microdeletion in region 7q11.23. The common critical region is about 1.6 Mb in size and contains more than 20 genes, including ELN (elastin) and LIMK1 (LIM domain kinase 1). Haploinsufficiency of ELN correlates with cardiovascular problems and is responsible for the SVAS phenotype of WBS patients. Deletions in region 7q11.23 are a consequence of non-allelic homologous recombination between low copy repeat elements flanking the WBS deleted region. Most WBS cases are sporadic, only few cases are transmitted vertically.
XL Williams-Beuren detects deletions in the long arm of chromosome 7. The orange labeled probe hybridizes to the ELN locus at 7q11.23. The green labeled probe hybridizes to a specific locus at 7p11.2 and functions as a reference probe.