Probes Newsletter 02/2022
Newsletter 02/2022
XCyting New Haematological Probes...
We are very pleased to announce four new XCyting locus-specific probes expanding our hematology and oncology portfolio. XL t(5;11) NSD1/NUP98 DF is designed to detect the cytogenetically cryptic translocation t(5;11)(q35;p15) resulting in NUP98::NSD1 gene fusion observed in pediatric acute myeloid leukemia (AML). XL ETV6 BA is a further developed version of our XL ETV6 (D-5073-100-OG). It features a new partially ETV6 gene covering design adapted to actualized customer demands. XL PAX5 BA is designed to detect PAX5 locus involving rearrangements. They have been described in childhood B-cell acute lymphoblastic leukemia (ALL), where ETV6, NOL4L, AUTS2, CBFA2T3, JAK2, and ZCCHC7 are the most frequent translocation partners. XL 5p15/21q22 is intended for non-centromeric simultaneous detection of trisomy 5 and 21. This is considered to be the most frequent combination among prognostic factors in multiple myeloma (MM).
XL t(5;11) NSD1/NUP98 DF
Clinical Applications: AML
Acute myeloid leukemia (AML) is a rare, heterogenic disease whose prognosis varies widely, depending on several factors such as chromosomal abnormalities. Conventional cytogenetics can detect structural and numerical cytogenetic abnormalities in about 50% patients with AML. However, products from cryptic translocations, loss of chromosome material or certain fusion genes, such as t(5;11)(q35;p15) NUP98::NSD1, can only be reliably detected using FISH or molecular genetic approaches as RT-PCR technique. NUP98 (Nucleoporin 98) located at 11p15.4 encodes a protein of the nucleopore complex. So far, more than 30 different fusion partner genes of NUP98 have been identified in various leukemias. The leukemogenesis seems to be mediated by changes in chromatin structure and gene expression. NSD1 (nuclear receptor binding SET domain protein 1) located at 5q35.3 was shown to be the most frequent NUP98 fusion partner gene in pediatric AML. NSD1 is discussed to function as a transcriptional coactivator and also as a corepressor. The chimeric protein, resulting from the fusion between the N-terminal part of NUP98 including phenylalanine-glycine (FG) repeats and the C-terminal part of NSD1 induces AML in vivo and enhances the expression of HOXA and HOXB. The frequency of NUP98::NSD1 translocations in AML is low and age-dependent, with a higher frequency in younger ages than in adults. For both, pediatric and adult NUP98::NSD1-positive AML patients, the prognosis is poor and often associated with primary resistance to chemotherapy. An association with FLT3 -ITD (internal tandem duplications), and/or WT1 mutations is reported in NUP98::NSD1 positive cases, supporting the hypothesis of a multistep AML pathogenesis.
XL t(5;11) NSD1/NUP98 DF consists of a green-labeled probe hybridizing to the NSD1 gene region at 5q35.2-35.3 and an orange-labeled probe hybridizing to the NUP98 gene region at 11p15.4.



XL ETV6 BA
Clinical Applications: ALL, AML, CML
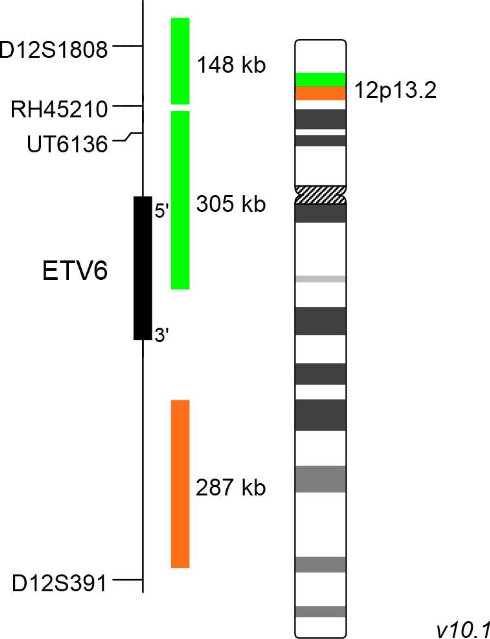
The ETV6 gene (ETS variant gene 6) located on 12p13.2 codes for a transcription factor which is involved in a variety of rearrangements. Several potential mechanisms of ETV6-mediated leukemogenesis are discussed including deletions and translocations. Numerous translocations and partner genes have been identified so far. The mechanisms well described are fusion to a tyrosine kinase resulting in a constitutively active tyrosine kinase and fusions to transcription factors driving aberrant expression of target genes. The frequent translocation t(12;21)(p13;q22) results in the formation of the chimeric transcription factor ETV6::RUNX1 which can be identified in about 25% of childhood B-cell acute lymphoblastic leukemia (B-ALL) cases. The t(5;12)(q33;p13) translocation fuses ETV6 to the receptor tyrosine kinase PDGFRB (ETV6::PDGFRB). The t(9;12)(q34;p13) ETV6::ABL1 has been included in the 5th edition of the WHO Classification of Haematolymphoid Tumours in the disease type myeloid/lymphoid neoplasms with eosinophilia and tyrosine kinase gene fusions (MLN-TK). The study by Haferlach et al confirms the variety of ETV6 rearrangements in acute myeloid leukemia (AML), myelodysplastic neoplasms (MDS), and MPNs, which have been shown to be associated with other genetic events. The recurrent translocation t(7;12)(q36;p13) can be identified in pediatric acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) and is resulting in the fusion gene MNX1::ETV6.
XL ETV6 BA is a further developed version of XL ETV6 (D-5073-100-OG), featuring a new, partially ETV6 gene covering design, extending into the 5´-region of the ETV6 gene.
XL ETV6 BA consists of an orange-labeled probe hybridizing proximal to the ETV6 gene region at 12p13.2 and a green-labeled probe hybridizing distal to the ETV6 gene region at 12p13.2 extending into the 5´gene region.


XL PAX5 BA
Clinical Applications: ALL
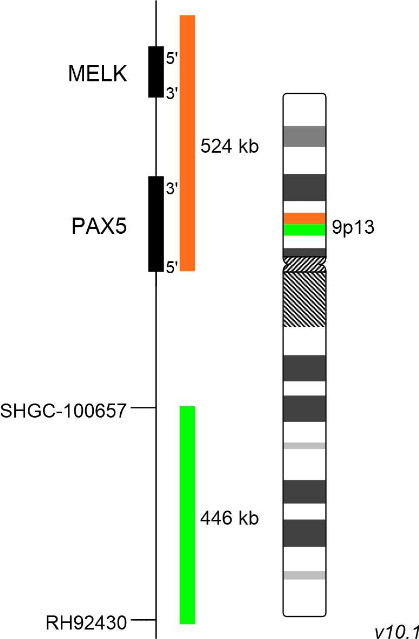
PAX5 (paired box 5), also known as B-cell specific activator protein (BSAP), is located on 9p13 and is a member of the paired box (PAX) family of transcription factors. The central characteristic is a highly conserved DNA-binding motif (paired box). PAX5 is a master regulator of B cell identity and development. It is expressed from early pro-B stage until final plasmacytic differentiation playing a dual role by activating B-cell commitment genes while repressing non–B-lineage genes. PAX5 alterations in B-cell acute lymphoblastic leukemia (B-ALL) include deletions of the whole gene, focal deletions, sequence mutations, intragenic amplification, and rearrangements. PAX5 rearrangements occurred at an incidence of about 2.5% in childhood B-ALL (Nebral et al) and PAX5 internal rearrangements are observed in 21% of B-ALL patients harboring 9p abnormalities (Coyaud et al). Two B-ALL subtypes, PAX5P80R and PAX5-altered (PAX5alt) are defined by different gene expression profiles and differential PAX5 alterations and considered as potential novel entities in the 5th edition of the World Health Organization (WHO) Classification of Haematolymphoid Tumours (2022). PAX5alt is characterized by various PAX5 alterations including chromosomal rearrangements resulting in dysregulated expression of PAX5 and subsequent transcriptional targets. Until 2019, 24 fusion partner genes of PAX5 were found, the most frequent being ETV6, NOL4L, AUTS2, CBFA2T3. PAX5::ETV6 results from dic(9;12)(p13;p13) which is associated with early suppression of B-cell differentiation. In PAX5::IGH/ t(9;14)(p13;q32), identified in a subset of aggressive B-cell non-Hodgkin lymphoma, the intronic Eµ enhancer of the IGH locus is juxtaposed next to the upstream promoter of PAX5, resulting in increased PAX5 expression.
XL PAX5 BA consists of an orange-labeled probe hybridizing to the PAX5 gene and distal gene regions at 9p13 and a green-labeled probe hybridizing proximal to the PAX5 gene region at 9p13.


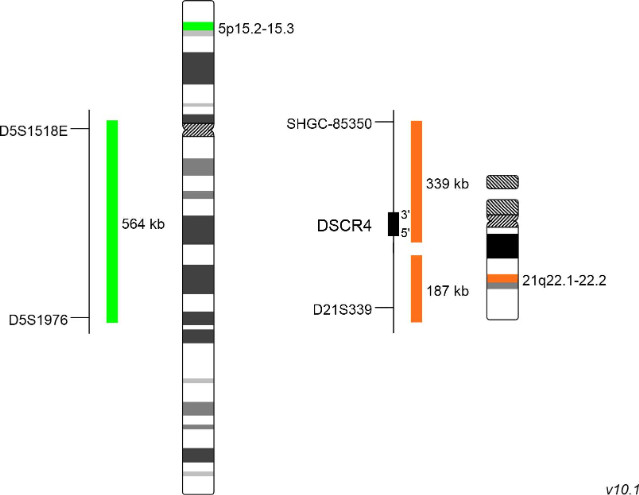
XL 5p15/21q22
Clinical Applications: MM
Multiple myeloma (MM) is a malignant disorder of plasma cells representing the second most prevalent hematologic malignancy, which is characterized by a wide heterogeneity of disease progression. While some of the MM patients stay alive more than 10 years after first diagnosis, others die within a few months. Chromosomal abnormalities in tumor plasma cells are of great significance among all prognostic factors analyzed in MM. To address the significant role of the different factors, the International Myeloma Working Group has recommended the implementation of these well studied chromosomal aberrations in the revised International Staging System (R-ISS) for MM. While del(17p), t(4;14), and t(14;16) are known to be among the high-risk factors in MM, trisomic MM is generally associated with a favorable outcome. More than half of MM cases diagnosed have a hyperdiploid karyotype, characterized by gains of the odd chromosomes.
While trisomy 3 or 5 significantly improves overall survival in MM patients, trisomy 21 has the adverse effect. The study of Perrot et al analyzed the combination frequency and prognostic impact of the factors del(17p), t(4;14), del(1p32), 1q21 gain, and trisomies 3, 5, and 21 within a big cohort and found out that the most frequent combination was the one between trisomy 5 and 21 among the aberrations examined.
The combined XL 5p15/21q22 fluorescence in situ hybridization (FISH) probe is a helpful aid for diagnosis to rapidly detect trisomy 5 and, or 21 in suspected or confirmed MM cases.
XL 5p15/21q22 consists of a green-labeled probe hybridizing to a region at 5p15.2-15.3 and an orange-labeled probe hybridizing to the DSCR4 gene region at 21q22.1-22.2.